E-FTMap Help
Welcome to E-FTMap! This page offers support for each facet of the E-FTMap webserver. Use the links below to jump to your topic of interest.
Contents
- Getting Started
- Job Submission
- Monitoring Jobs
- Viewing job results
- Advanced Option: Creating custom probe sets
- Tutorial: Submit a job by specifying binding site residues
- Tutorial: Submit a job using a computationally predicted binding site
Getting Started with E-FTMap
In order to view and submit jobs to E-FTMap, you need to login to your account.
If you don't have an E-FTMap account yet, please sign up. When making your account you must provide your First Name, Short Name, Email Address, and a Password. Optionally, you can also provide your Affiliation and Organization. Click the Sign Up button when you are done.
If you are a returning user and have forgotten your password, click Forgot password? to reset your password.
E-FTMap is free for academic and governmental use. If you are unable to make an account with your e-mail, please contact us. If you wish to use E-FTMap for commercial purposes, contact Acpharis at https://acpharis.com.
Job Submission
Once you have logged in, you can submit jobs with E-FTMap. A user can submit up to 20 jobs in a 24 hour period.
To submit a job, navigate to the Submit page, shown here:
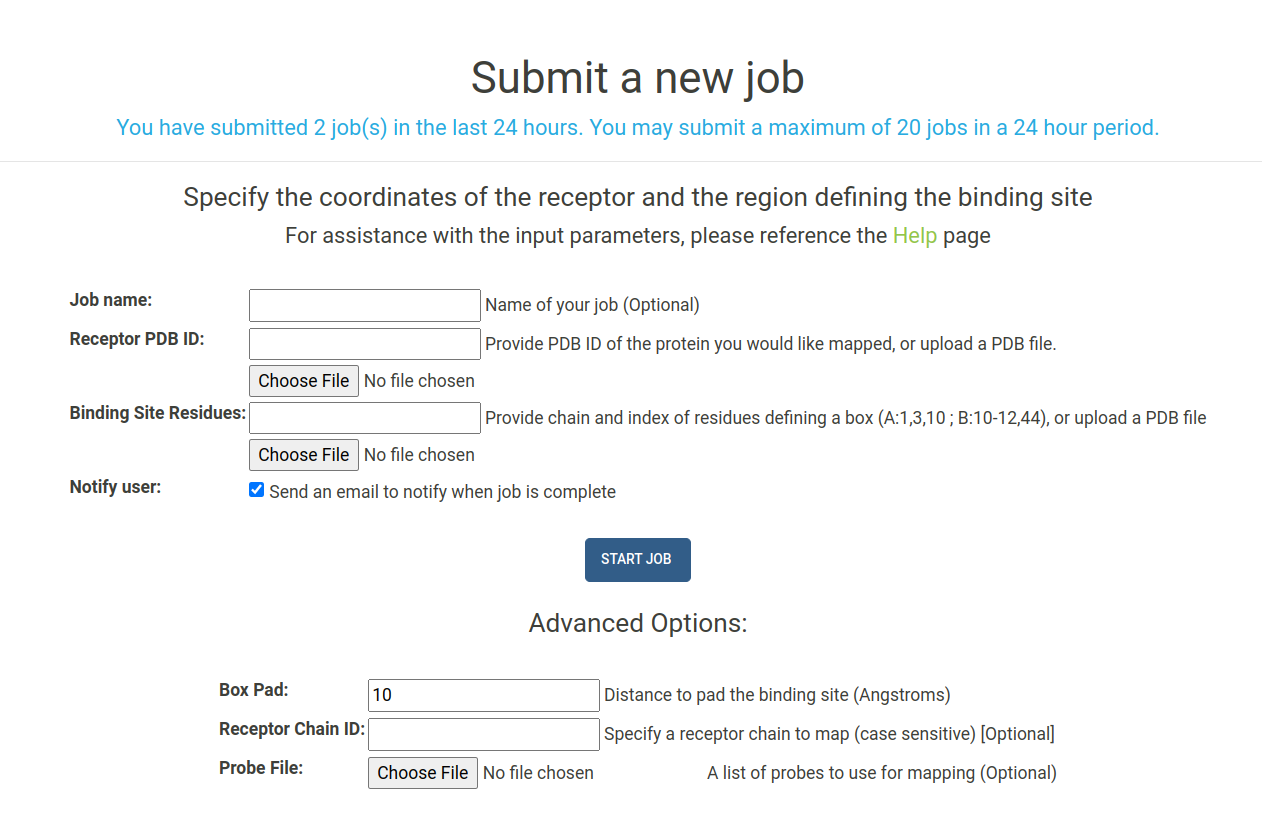
A number of arguments can be given when submitting your job. Each input parameter is outlined below:
- Job Name : (Optional) Provide a name for your job submission. If you do not provide a name it will automatically be created.
- Receptor PDB ID : Select the receptor by entering the four digit PDB ID of the protein you would like to map. All structures are downloaded from the RCSB. Alternatively, you can upload a PDB file for the receptor you would like to map.
-
Binding Site Residues : Provide a list of residues in the receptor to define the binding site. The chain of
the binding site residues must be provided, followed by a colon ,followed by a list of
comma separated residue index numbers. Examples of binding site inputs are listed below:
-
To select a binding site defined by residues 1, 3, and 10-13 in chain A of the
receptor provide the input:
ex 1. "A:1,3,10-13" -
To select a binding site at the interface of chains A and B, you can denote residue
selections within each chain using a semicolon:
ex 2. "A:1,3,10-13;B:10-12,44" - Alternatively, upload the atomic coordinates of a molecule that is in your binding site in PDB format.
-
To select a binding site defined by residues 1, 3, and 10-13 in chain A of the
receptor provide the input:
- Notify User : Check this box to receive an email from E-FTMap when your job is complete
Additional advanced options can also be provided:
-
Box Pad : The distance by which you wish to "pad" your binding site in the x,
y , and z directions. By default, E-FTMap will create a box that extends 10 angstroms beyond the maximum and minimum x,y , and z coordinates in the binding site that is defined. - Receptor Chain ID : Specify a specific chain in the receptor you wish to map. If a multi-chain protein is selected, only this chain will be used as the receptor in mapping calculations.
- Probe File : Upload a text file specifying a custom probe set to use in the mapping calculation. If left blank, the default E-FTMap probe set will be used. Read more about how to create a probe file in the Creating custom probe sets section.
Monitoring jobs
To check the status of your jobs, navigate to the My Jobs page, shown below:
All jobs are run in the order they are submitted. The runtime for your job is dependent on the availability of computational resources, the size of the receptor being mapped, and the number of probes you use in your mapping calculation.
A list of the jobs you submitted can be viewed in the "Jobs in progress" table. Jobs that have successfully finished running are listed in the "Completed jobs" table. Each job will have one of the following statuses:
- PDB Download : The receptor coordinates are being downloaded the RCSB PDB.
- PDB Modification : The receptor PDB file is being modified so that it can be used for mapping. Modifications include the removal of all nonstandard residues, and the selection of receptor chains specified by the user.
- Gen Job Pkg : All input files are being processed for submission to Boston University's Shared Computing Cluster (SCC)
- Scc Submit : The job is being submitted to SCC
- Scc Running : The job has successfully been submitted to the SCC and the webserver is waiting to receive results.
- Scc Results : Results from the mapping calculation are being unpacked
- Complete : The job has been completed, and results can now be viewed and downloaded.
- Error : An error has occurred with your job, and it will no longer be run.
- Deleted : The job has been deleted. Jobs are deleted automatically after 30 days, or manually by the user.
Viewing job results
By clicking the name of one of your jobs on the My Jobs page, you can access the results page for your job. Here you can view a a detailed list describing the status of the job, the type of input used for the receptor coordinates, the type of input used to define the binding site box, the chains used for mapping, the date and time when the job was created, and the date and time when the job was last modified.
Users can restart or delete a job at any time using the Delete Job or Restart Job buttons located in the top right of the page.
Prior to the completion of a job, the NGL Molecular Viewer can be used to view the structure of the input file that is being mapped. When a job is completed, E-FTMap results can be viewed in the browser using the E-FTMap Results button.
A screenshot of the results page can be seen below. When viewing results with the E-FTMap Results button, a legend is displayed to the right of the viewer, showing the colors of each atomic consensus cluster, the rank of each atomic consensus cluster, and the size (number of probe atoms) in each atomic consensus cluster. Pharmacophore features are shown in the ball and stick representation, and the receptor is shown in a grey cartoon representation.
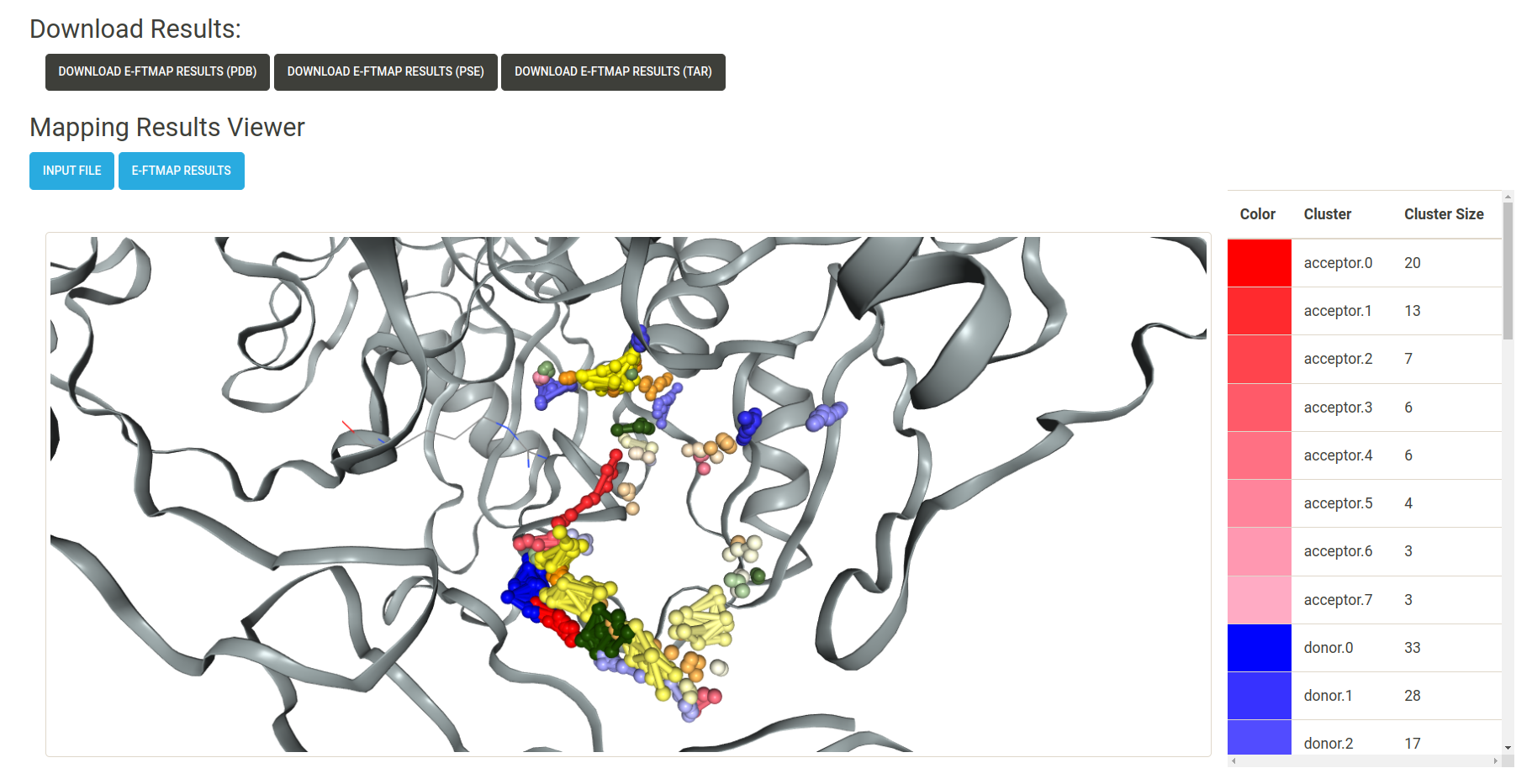
When a job is completed, the following download buttons are displayed on the results page:
- Download E-FTMap Results (pdb) : Download a PDB file containing the coordinates of the receptor and atomic consensus clusters found by E-FTMap. Each atomic consensus cluster is separated by a HEADER entry in the PDB file.
- Download E-FTMap Results (pse) : Download a Pymol Session (.pse) file which displays the results from the E-FTMap calculation. This file has been formatted to allow for an easy visualization of the mapping results, in the style of the figures presented in the E-FTMap publication. Options to visualize the top 3, top 5, and top 10 pharmacophore features are saved as "scenes" in the Pymol session. Additional scenes can be used to view atomic consensus sites for each of the pharmacophore atom types.
-
Download E-FTMap Results (tar) : A .tar file containing additional information from the mapping
results. The contents of this file include:
- The inputs/ directory : Contains pdb files for the receptor (rec.pdb) and binding site (rec.box.pdb)
- The cluster_sdfs/ directory : Contains sdf files for the probes which have been included in each atomic consensus cluster
- recrun_pharmacophore_clusters.pdb : Coordinates of the receptor and atomic consensus clusters generated by E-FTMap calculations in PDB format
- recrun_pharmacophore_visual.pse : A visualization of the E-FTMap results in PSE format
- cluster_energy_log.txt : A log file with the CHARMM energy for each probe in each cluster
We recommend viewing E-FTMap results using the PyMOL Session (.pse) file that is provided to visualize E-FTMap results, since it includes with number of features which ease the viewing experience. A screenshot of the PyMOL visualization can be seen below:
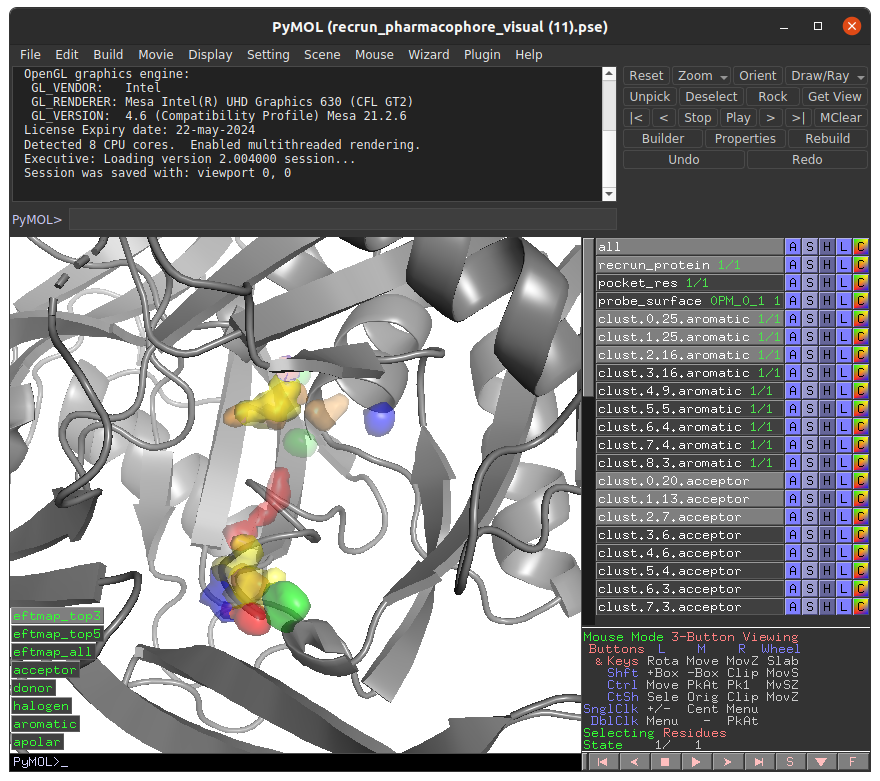
In this .pse file the these objects can be enabled to visualize the following:
- recrun_protein: The coordinates of the mapped receptor
- pocket_res: Receptor residues within 5A of the E-FTMap probes
- probe_surface: Regions on the receptor surface which E-FTMap probes accessed during the calculation
- clust.X.Y.aromatic: Aromatic atomic consensus site of rank X containing Y probe atoms
- clust.X.Y.acceptor: Hydrogen bond acceptor atomic consensus site of rank X containing Y probe atoms
- clust.X.Y.donor: Hydrogen bond donor atomic consensus site of rank X containing Y probe atoms
- clust.X.Y.apolar: Apolar atomic consensus site of rank X containing Y probe atoms
- clust.X.Y.halogen: Halogen atomic consensus site of rank X containing Y probe atoms
- eftmap_top3: Display the top 3 ranked atomic consensus sites for each atom type
- eftmap_top5: Display the top 5 ranked atomic consensus sites for each atom type
- eftmap_all: Display all ranked atomic consensus sites for each atom type
- acceptor: Display all atomic consensus sites for the acceptor atom type
- donor: Display all atomic consensus sites for the donor atom type
- halogen: Display all atomic consensus sites for the halogen atom type
- aromatic: Display all atomic consensus sites for the aromatic atom type
- apolar Display all atomic consensus sites for the apolar atom type
Advanced Option: Creating custom probe sets
If you wish to create a custom set of probes to use in mapping calculations, you can generate a probe text file. Each line in the probe file must have three whitespace-separated columns containing the following information:
- Resdiue Name : A three character residue name unique to the probe
-
SMILES String : A smiles string for the probe you wish to use. Note that the protonation states of heteroatoms must be
explicitly defined:
ex 1. Define ethanol as CCO[H] instead of CCOex 2. Define charged carboxylate as CC(=O)[O-] instead of CC(=O)O - Probe Set Name : The name of the set the probe belongs to. E-FTMap carries out independent mapping calculations for each probe set. We recommend grouping probes with similar chemical properties into the same set to achieve the best results. (i.e. A set of aliphatic probes, a set of aromatic probes, etc.)
Download a template text file : A template text file used to generate the default E-FTMap probe set, which contains 28 distinct probe sets, can be downloaded here. It can be modified to include a custom user-defined probe set.
Please note that increasing the number of probes will increase the runtime of your job. If a job takes more than 30 hours to complete, it will automatically be timed out. To reduce runtimes, we recommend including no more than 16 probe molecules in each probe set.
Tutorial: Submit a job using binding site residues
To submit a job using binding site residues as the input, you must provide the chain and index numbers of the residues in the binding site. In this tutorial, we will select binding residues within the ADP binding site of human NUDT5, and supply these residues as inputs in the E-FTMap calculation. For the sake of this tutorial, let's assume that we have identified two residues in the receptor that are important for ligand binding. In the PyMOL visualization of the receptor we plan to map (PDB ID: 6GRU), we see TRP 46 in chain A of the receptor (green), and ARG 51 in chain B of the receptor (cyan) are displayed:
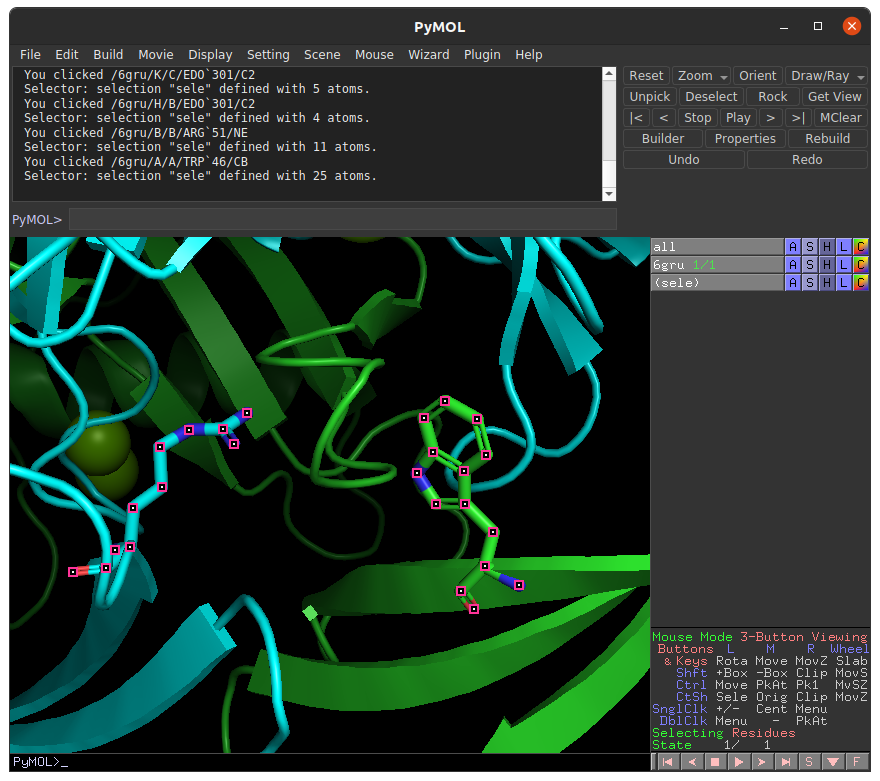
To supply this information to our E-FTMap job, we can navigate to the Submit page. Under the Receptor PDB ID field, provide the four-digit PDB accession code for the structure you want to map. In this example we input the PDB ID 6GRU.
Next, we must specify which residues are a part of the binding site. E-FTMap allows users to specify the chain and index of multiple residues that are in or near the binding site. Since we know that TRP 46 in chain A of the receptor will be part of the binding site, we can provide this information in the Binding Site Residues field by typing "A:46;B:51". This specified that in the 6GRU, we wish to specify residue 46 from chain A, and residue 51 from chain B as part of the binding site we wish to map. For more detailed descriptions of residues in the binding site, please refer to the directions given in the Job Submission section of the Help page.
When a user supplies a selection of binding site residues to E-FTMap, the coordinates of all residues in the selection are used to create a cubic box containing all binding site residues. This box will be used to restrict E-FTMap calculations to the local region in which these residues are contained.
Once all inputs are provided, click the "Start Job" button to begin your E-FTMap calculation:
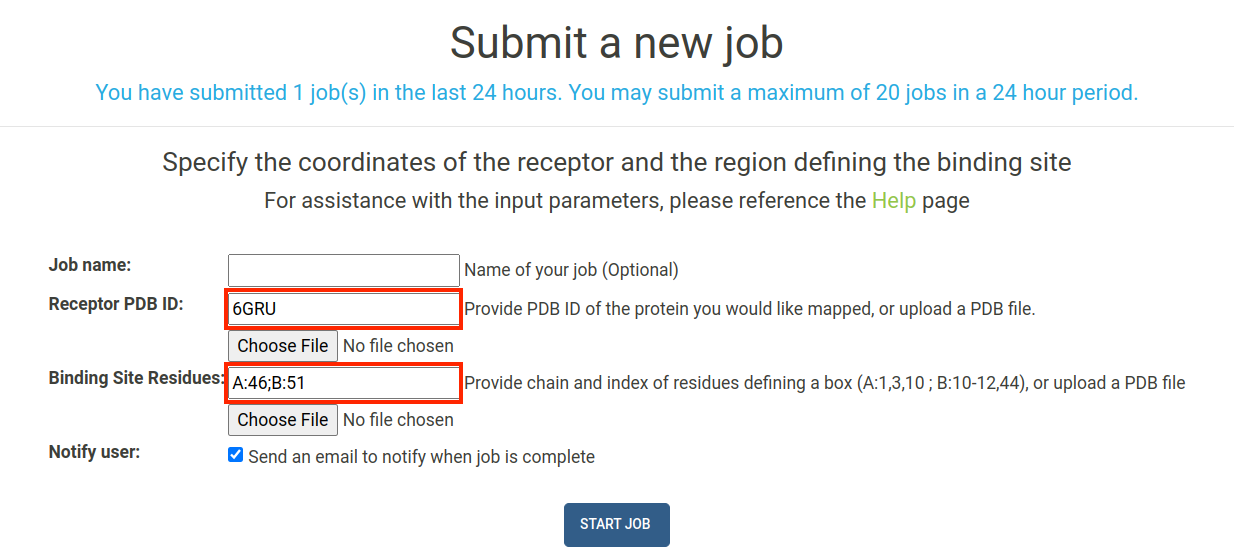
IMPORTANT: The residue selection you provide must be specific to the receptor you are mapping! Residue numbering is often inconsistent between multiple crystal structures of the same protein in the PDB, so we recommend visualizing your binding site residues in PyMOL to confirm that your selection is correct!
Tutorial: Submit a job using a computationally predicted binding site
In the event that you wish to run E-FTMap, but do not have an experimentally validated binding site, you can use computational tools to predict where the ligand binding site will be. In this tutorial, we will use the FTSite webserver to generate a predicted binding sites on an apo crystal structure of human NUDT5 (PDB ID: 6GRU). Then, the predicted binding site will be downloaded from FTSite, and uploaded to E-FTMap to be used in our job.
To begin, we navigate to the FTSite webserver and submit a job using the PDB ID 6GRU. Once the job is complete we can download the results and view them in PyMOL:
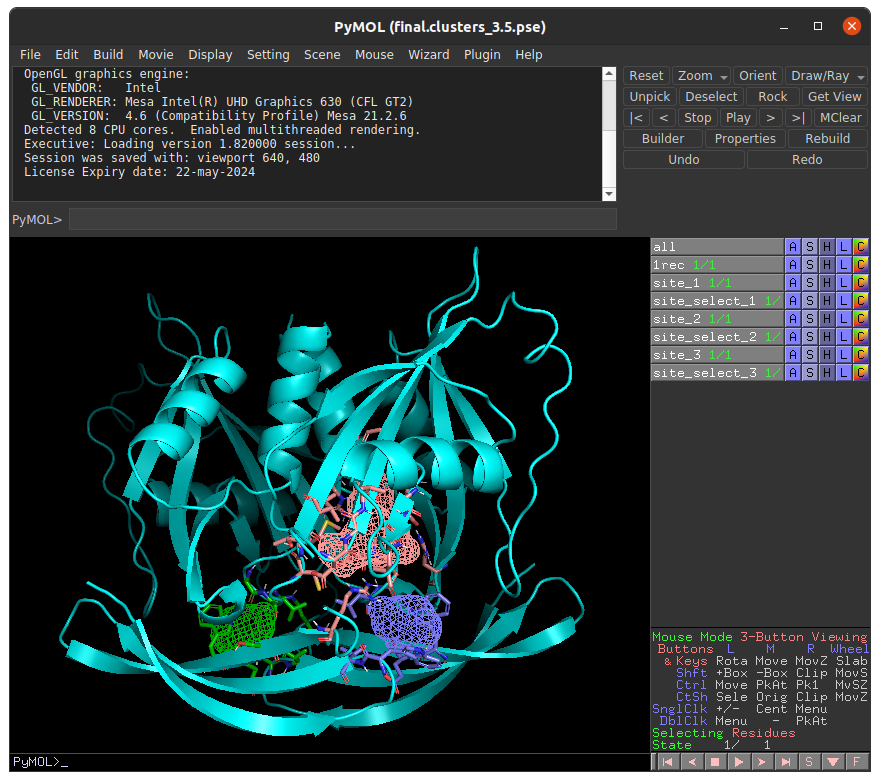
In this example, we will choose to investigate to top ranked binding site prediction (labeled as site_1) in with E-FTMap. In order to prepare the inputs for E-FTMap, we must save the coordinates of the receptor and binding site probes in separate PDB files. To do this, run the following commands in PyMOL:
- To save the receptor: save receptor.pdb, 1rec
- To save the binding site prediction: save site_1.pdb, site_1
To use these files in E-FTMap, navigate to the Submit page. To upload the receptor, click the "Choose File" button under the Receptor PDB ID field, and upload receptor.pdb. To upload the binding site coordinates, click the "Choose File" button under the Binding Site Residues field, and upload site_1.pdb. After uploading each file, click "Start Job" to begin your E-FTMap calculation:
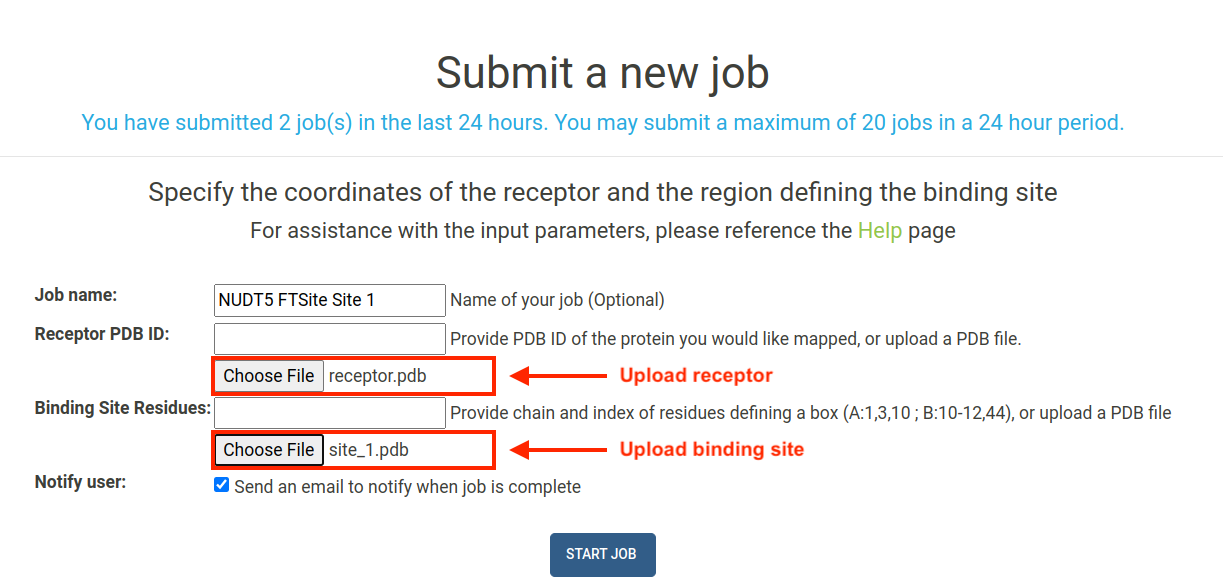
IMPORTANT: If you are uploading custom files for the receptor and binding site, please ensure that the coordinates of the receptor are properly aligned with the predicted coordinates of the binding site.